
Создание и характеристика новой трансгенной модели бокового амиотрофического склероза, основанной на нейроспецифической экспрессии патогенной формы белка FUS
Боковой амиотрофический склероз (БАС) является третьим по частоте встречаемости нейродегенеративным заболеванием в мире с частотой возникновения 1,5-2,5 на 100 000 в год. Непосредственные молекулярные механизмы, лежащие в основе патологических процессов, ведущих к селективной гибели двигательных нейронов, остаются недостаточно изученными. Это является главной причиной отсутствия прогресса в разработке эффективных методов лечения бокового амиотрофического склероза, тяжелого летального заболевания нервной системы. Актуальной задачей до настоящего времени остается создание адекватных животных моделей, в которых возможно воспроизведение основных характеристик патогенеза БАС и выявление ключевых молекулярных мишеней для разработки патогенетической терапии данного заболевания. Полученные в различных лабораториях данные о возможном участии ДНК/РНК-связывающего белка FUS в патогенезе БАС послужили основой для создания гипотезы о его важной роли в патологических механизмах избирательного повреждения двигательных нейронов. Эта гипотеза требовала прямых экспериментальных доказательств. Mоделирование FUSопатии, которая приводит к развитию у трансгенных животных фенотипа, воспроизведящего основные патогенетические признаки БАС, является наиболее убедительным методом доказательства ключевой роли белка FUS в инициации и прогрессии патологического процесса, характерного для данного заболевания.
В нашей лаборатории была создана первая модельная линия трансгенных мышей, в которых эктопическая экспрессия укороченной формы белка FUS человека 1-359 позволила воспроизвести классическую FUSопатию. Впервые экспериментально доказано, что прогрессия FUSопатии у Thy-1/FUS 1-359 мышей вызывает развитие нейродегенеративного процесса с селективной потерей двигательных нейронов, то есть путем экспрессии аберрантной формы белка FUS, без участия каких-либо других патогенных факторов, удалось воспроизвести в лабораторных мышах основные патогенетические признаки БАС. Получены новые данные, позволяющие рассматривать образование аберантной укороченной формы белка FUS в качестве оригинальной молекулярной мишени для разработки патофизиологической терапии определенных форм БАС.
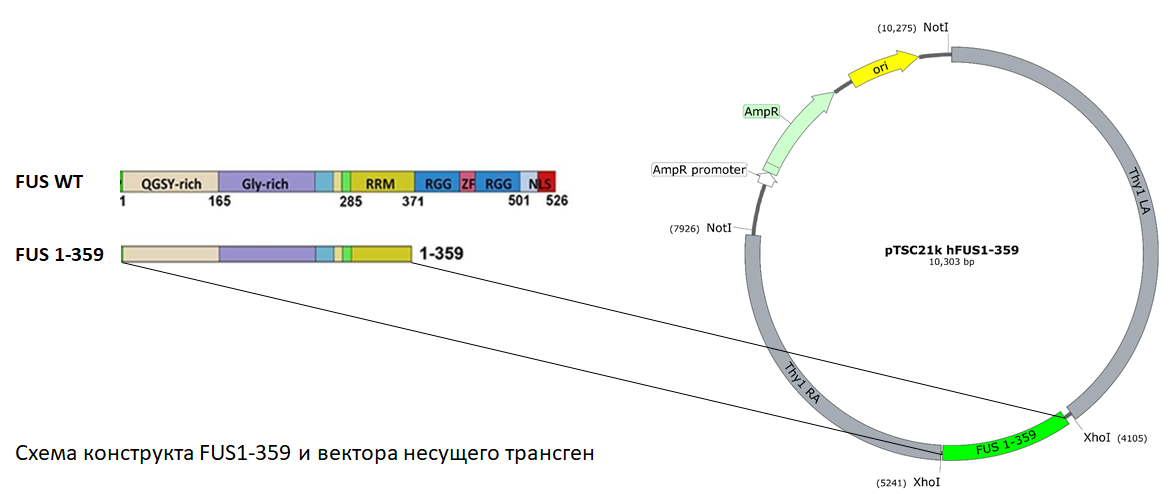
При исследовании механизмов протеинопатий, ассоциированных с БАС, был описан новый тип молекулярно-клеточной патологии, обусловленный нарушением функции ДНК/РНК-связывающего белка FUS и его неспособностью формировать физиологически активные и легко диссоциирующие комплексы с РНК. Вместо этого наблюдалось образование стабильных внутриклеточных включений, не содержащих РНК, в составе которых был депонирован агрегированный белок FUS с измененной конформацией. При этом менялась компартментализация FUS и накопление его в цитоплазме, что указывало на возможную роль этого белка FUS в патогенезе нейродегенерации двигательных нейронов. Важным аргументом в пользу того, что ключевым событием в патогенезе БАС могут являться нарушения функции FUS, служили данные генетического анализа пациентов с наследственными формами с мутациями в этом гене. Однако прямые доказательства причинной роли белка FUS в механизмах специфической дегенерации двигательных нейронов отсутствовали. Более того, анализ созданных в различных лабораториях FUS трансгенных животных показал, что не всякий тип нарушения функции FUS приводит к развитию нейродегенеративного процесса. Выработка экспериментального подхода в данном диссертационном исследовании базировалась на выдвинутой нами гипотезе, согласно которой специфическое изменение структуры белковой молекулы FUS по типу патогенной формы сходного белка TDP-43, обнаруживаемой в составе патогенетических включений в аутопсийном материале больных с БАС, позволит воспроизвести FUSопатию в трансгенных мышах, которая должна привести к развитию нейродегенеративного процесса в двигательных нейронах. Нами была отобрана модифицированная форма белка FUS 1-359, у которой отсутствовал сигнал ядерной локализации и были повреждены РНК распознающий и РНК связывающий домены, что вело к нарушению его взаимодействия с РНК и перераспределению белка в клетке с накоплением в цитоплазме. Следует отметить, что в последовательностях гена, кодирующих эти участки белка FUS, было обнаружено наибольшее число мутаций, ассоциированых с наследственными формами БАС.
Существенным преимуществом полученной модели являлось развитие патологического фенотипа при исключительно низком содержании трансгенного белка человека, которое было достаточным для инициации и развития нейродегенеративного процесса. Животные рождались без внешних признаков патологии: они нормально развивались, были фертильны и оставляли жизнеспособное потомство. Первые признаки, наиболее часто выражавшиеся в прогрессирующих односторонних парезах задних и передних конечностей, отмечались у взрослых половозрелых животных, начиная с возраста 10-12 недель. Неврологическая симптоматика развивалась стремительно на фоне хорошего общего здоровья мышей, модельное заболевание характеризовалось быстрой прогрессией и часто даже инструметальными методами не удавалось выявить у животных отклонения, предшествующие переходу из пресимптоматической в симптоматическую стадию. Не позднее, чем через две недели после выявления первых симптомов заболевание переходило в терминальную стадию с тетраплегией и к возрасту 25 недель все трансгенные животные погибали.
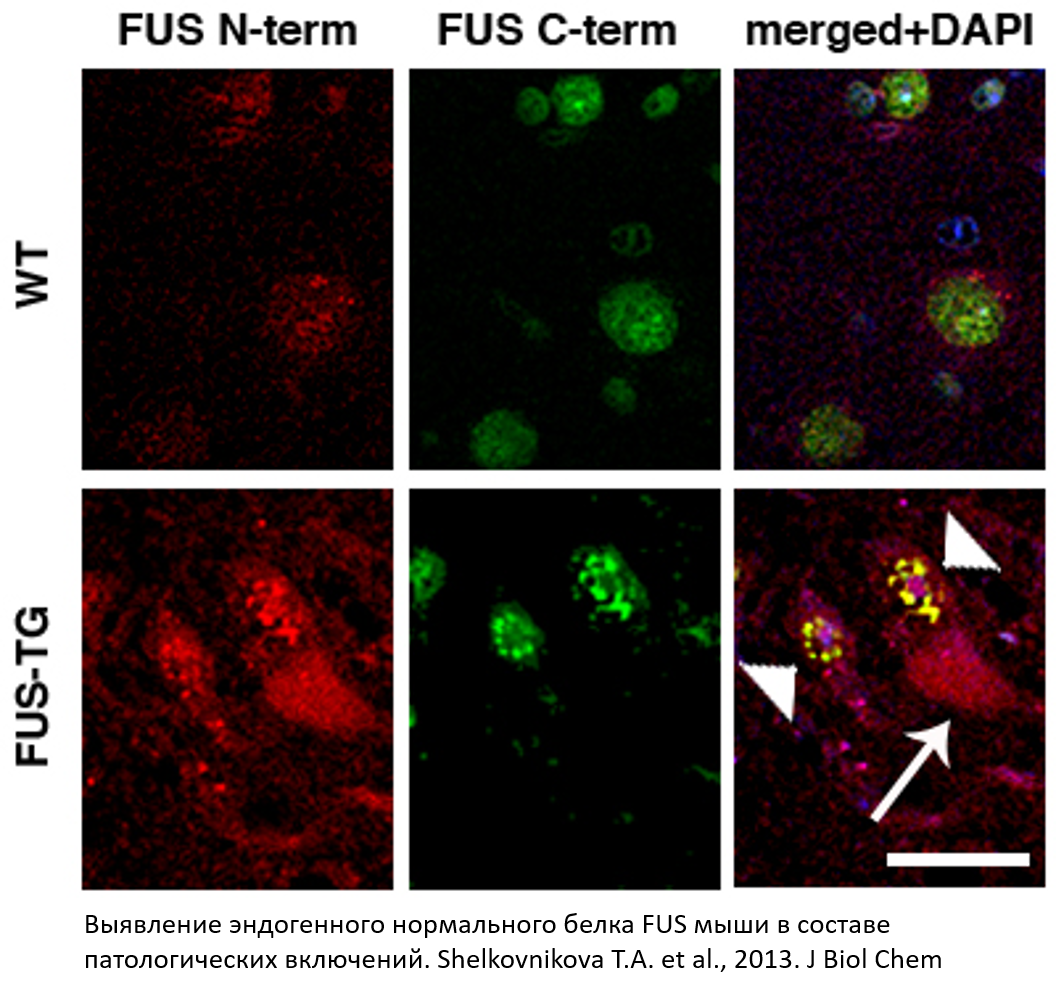
Важным элементом доказательства нашей гипотезы о ключевой роли FUSопатии в развитии специфического нейродегенеративного процесса с преимущественным поражением двигательных нейронов явился патогистологический анализ FUS-реактивных включений в спинном мозге Thy-1/FUS 1-359 мышей, выявивший присутствие в составе включений эндогенного полноразмерного белка FUS мыши. Таким образом было показано, что аберантная форма FUS (1-359) человека способна индуцировать агрегацию эндогенного FUS мыши и увлекать его в состав патогистологических депозитов, т.е. инициировать протеинопатию. Исключение белка в результате индуцированной агрегации из нормального метаболизма и его последующая функциональная недостаточность приводит к необратимым патологическим изменениям.